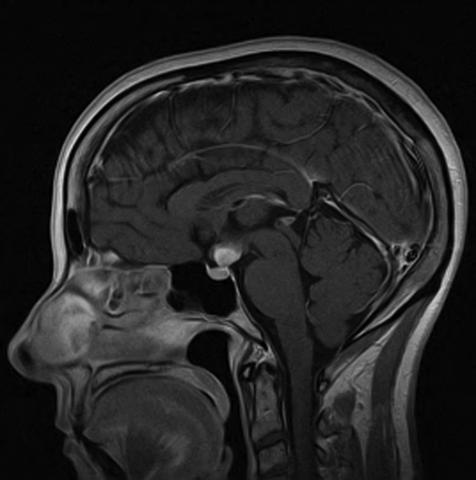
A 55-year-old woman presented with polyuria, nocturia, and thirst-induced polydipsia that developed insidiously over a six-month period. She was seen by her primary physician, who suspected and confirmed a diagnosis of diabetes insipidus. She was treated with DDAVP that provided excellent symptom control. Laboratory studies reflected normal anterior pituitary function. She was otherwise healthy. Physical examination was unrevealing. Magnetic resonance imaging of the sella illustrated a 1.5-cm mass in the region of the hypothalamus.
The differential diagnosis for such a lesion includes neurosarcoidosis, Langerhans cell histiocytosis, germinoma, hypothalamic glioma, metastatic cancer, fungal infection, and tuberculosis. The patient underwent surgery to permit biopsy of the lesion. Findings were classic for a diagnosis of Langerhans cell histiocytosis.
Langerhans cell histiocytosis is a disorder that represents a clonal proliferation of cells that are derived from the bone marrow and resemble the epidermal dendritic cells that present androgens to the immune system. This disorder has been known as Histiocytosis X, Hand-Schuller- Christian Disease, Abt-Letterer- Siwe Disease, and eosinophilic granuloma. The peak incidence of the disorder is between 5 and 10 years of age. It occurs extremely rarely in adults with the frequency estimated to be one in 560,000 adult persons. The patient presented is, to my knowledge, the oldest individual seen in our institution with the disorder.
Langerhans cell histiocytosis can present with mono-ostotic or polyostotic bone involvement alone. The affected bones are usually swollen and painful. Pathological fractures may occur. It may involve the in fundibulum, resulting in diabetes insipidus in varying degrees of hypopituitarism, and the skin, particularly of the scalp and the ear canals, as is often seen in children.
Rarely, the disease may involve multiple body systems, including the bone marrow, leading to pancytopenia; the lungs, leading to restrictive lung disease; the liver; the spleen; and the lymph nodes, and can progress rapidly, leading to death in 50% of patients. This aggressive form of the disorder is most often seen in children under the age of 2 years but has been reported in adults. I have personally seen one similarly affected 35-year-old man who was originally thought to have multiple skull-based meningiomas, which proved to be histiocytomas, as one component of his multi-system illness.
There are no well-defined approaches to treatment of patients with Langerhans cell histiocytosis. Fortunately, most patients with limited disease do well and observation may be all that is required. Patients with mass effects in the region of the hypothalamus and pituitary may require debulking surgical procedures. Radiotherapy is usually employed for lesions that demonstrate a progressive increase in size during a period of follow-up.
Topical steroids may result in involution of the lesions in the skin and ear canals. Systemic chemotherapy employing agents such as vincristine, vinblastine, etoposide, methotrexate, thalidomide, and prednisone, alone or in combination, have been employed with variable levels of success. At present, there are no active protocols registered with the National Cancer Institute for the experimental treatment of Langerhans cell histiocytosis.